Note
Go to the end to download the full example code
Leontis-Westhof Nomenclature#
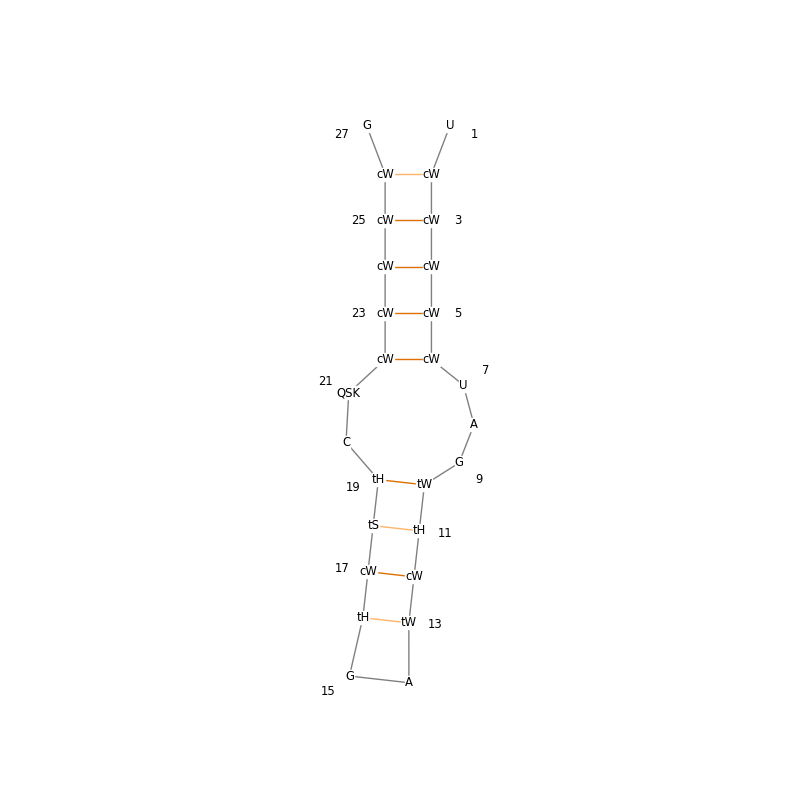
In this example we plot a secondary structure diagram annotated with Leontis-Westhof nomenclature [1] of the sarcin-ricin loop from E. coli (PDB ID: 6ZYB).
# Code source: Tom David Müller
# License: BSD 3 clause
from tempfile import gettempdir
import matplotlib.pyplot as plt
import numpy as np
import biotite
import biotite.database.rcsb as rcsb
import biotite.structure as struc
import biotite.structure.graphics as graphics
import biotite.structure.io.pdb as pdb
# Download the PDB file and read the structure
pdb_file_path = rcsb.fetch("6ZYB", "pdb", gettempdir())
pdb_file = pdb.PDBFile.read(pdb_file_path)
atom_array = pdb.get_structure(pdb_file)[0]
nucleotides = atom_array[struc.filter_nucleotides(atom_array)]
# Compute the base pairs and the Leontis-Westhof nomenclature
base_pairs = struc.base_pairs(nucleotides)
glycosidic_bonds = struc.base_pairs_glycosidic_bond(nucleotides, base_pairs)
edges = struc.base_pairs_edge(nucleotides, base_pairs)
base_pairs = struc.get_residue_positions(nucleotides, base_pairs.flatten()).reshape(
base_pairs.shape
)
# Get the one-letter-codes of the bases
base_labels = []
for base in struc.residue_iter(nucleotides):
base_labels.append(base.res_name[0])
# Color canonical Watson-Crick base pairs with a darker orange and
# non-canonical base pairs with a lighter orange
colors = np.full(base_pairs.shape[0], biotite.colors["brightorange"])
for i, (base1, base2) in enumerate(base_pairs):
name1 = base_labels[base1]
name2 = base_labels[base2]
if sorted([name1, name2]) in [["A", "U"], ["C", "G"]]:
colors[i] = biotite.colors["dimorange"]
# Use the base labels to indicate the Leontis-Westhof nomenclature
for bases, edge_types, orientation in zip(base_pairs, edges, glycosidic_bonds):
for base, edge in zip(bases, edge_types):
if orientation == 1:
annotation = "c"
else:
annotation = "t"
if edge == 1:
annotation += "W"
elif edge == 2:
annotation += "H"
else:
annotation += "S"
base_labels[base] = annotation
# Create a matplotlib pyplot
fig, ax = plt.subplots(figsize=(8.0, 8.0))
# Plot the secondary structure
graphics.plot_nucleotide_secondary_structure(
ax, base_labels, base_pairs, struc.get_residue_count(nucleotides), bond_color=colors
)
# Display the plot
plt.show()
/usr/share/miniconda3/envs/test/lib/python3.11/site-packages/biotite/structure/hbond.py:270: UserWarning: Input structure has no associated 'BondList', Hydrogen atoms bonded to donors are detected by distance
warnings.warn(
The sarcin-ricin loop is part of the 23s rRNA and is considered crucial to the ribosome‘s activity. The incorporation of the Leontis-Westhof nomenclature into the 2D-plot shows how the individual base pairs are oriented and how their glycosidic bonds are oriented relative to each other.
This visualization enables one to see a pattern that cannot be communicated through the 2D structure alone. The upper part of the sarcin-ricin loop consists of only cis (c) oriented glycosidic bonds. All bases interact through their Watson-Crick edge (W). On the other hand, the lower part of the sarcin ricin loop looks strikingly different. The glycosidic bonds are oriented in cis (c) and trans (t) orientation. The bases interact through all three edges: Watson-Crick (W), Hoogsteen (H), and Sugar (S).
Thus, it can be concluded that the upper part of the sarcin ricin loop represents a highly organized helix, while the lower part of the loop is comparatively unorganized.