Note
Go to the end to download the full example code
Contact sites of protein-DNA interaction#
This script identifies which amino acids of the phage 434 repressor are in contact with its corresponding DNA operator. In contact is defined as a pairwise atom distance below a given threshold (in this case 4.0 Å).
The identified contact residues are highlighted as sticks.
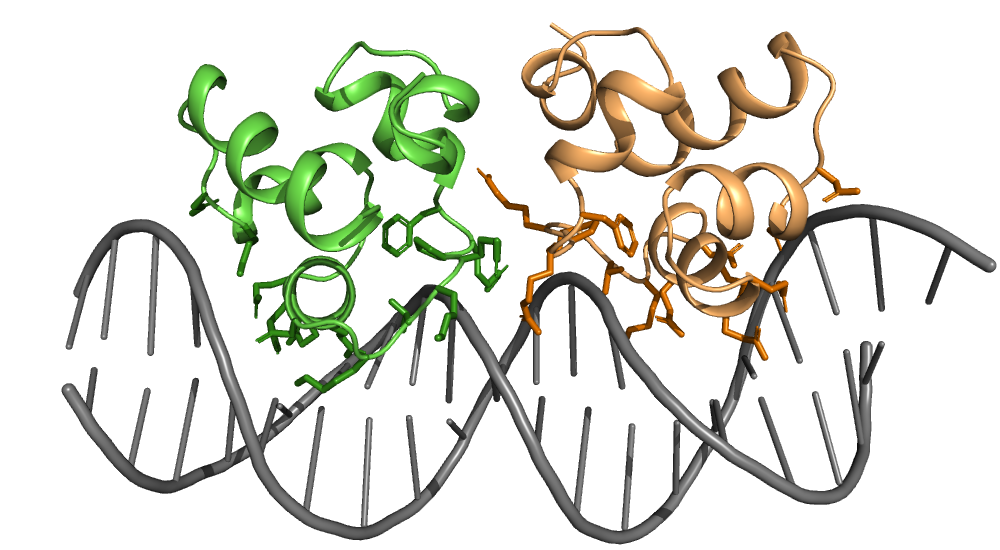
Residues in contact with DNA:
Asn16
Gln17
Gln28
Gln29
Glu32
Gln33
Asn36
Lys38
Thr39
Lys40
Arg41
Pro42
Arg43
Phe44
# Code source: Patrick Kunzmann
# License: BSD 3 clause
import numpy as np
import biotite.database.rcsb as rcsb
import biotite.structure as struc
import biotite.structure.io.pdbx as pdbx
# The maximum distance between an atom in the repressor and an atom in
# the DNA for them to be considered 'in contact'
THRESHOLD_DISTANCE = 4.0
# Fetch and load structure
pdbx_file = pdbx.BinaryCIFFile.read(rcsb.fetch("2or1", "bcif"))
structure = pdbx.get_structure(pdbx_file, model=1)
# Separate structure into the DNA and the two identical protein chains
dna = structure[np.isin(structure.chain_id, ["A", "B"]) & ~structure.hetero]
protein_l = structure[(structure.chain_id == "L") & ~structure.hetero]
protein_r = structure[(structure.chain_id == "R") & ~structure.hetero]
# Quick check if the two protein chains are really identical
assert len(struc.get_residues(protein_l)) == len(struc.get_residues(protein_r))
# Fast identification of contacts via a cell list:
# The cell list is initiliazed with the coordinates of the DNA
# and later provided with the atom coordinates of the two protein chains
cell_list = struc.CellList(dna, cell_size=THRESHOLD_DISTANCE)
# Sets to store the residue IDs of contact residues
# for each protein chain
id_set_l = set()
id_set_r = set()
for protein, res_id_set in zip((protein_l, protein_r), (id_set_l, id_set_r)):
# For each atom in the protein chain,
# find all atoms in the DNA that are in contact with it
contacts = cell_list.get_atoms(protein.coord, radius=THRESHOLD_DISTANCE)
# Only retain atoms in the protein with contact
# to at least one atom of the DNA
contact_indices = np.where((contacts != -1).any(axis=1))[0]
# Get residue IDs for the atoms in the protein
contact_res_ids = protein.res_id[contact_indices]
# Put the residue IDs into the set,
# duplicate IDs are automatically removed in this process
res_id_set.update(contact_res_ids)
# Only consider residues that show contacts in both peptide chains
# -> intersection of sets
common_ids = sorted(id_set_l & id_set_r)
# Print output
print("Residues in contact with DNA:")
print()
for res_id in common_ids:
res_name = protein_l.res_name[protein_l.res_id == res_id][0]
print(res_name.capitalize() + str(res_id))
# Visualization with PyMOL...