Note
Go to the end to download the full example code
Finding homologs of a gene in a genome#
In this example we use a combination of a KmerTable
and align_banded() to search a genome for homologs to a gene
from another organism.
Specifically we take the gene coding for the M1 RNA from
Escherichia coli BL21, an RNA subunit of the ribonuclease P,
and try to find the counterpart in the genome of Salmonella enterica.
This approach has a high performance compared to align_optimal()
and is similar to the method used by software like BLAST.
At first the sequences for the M1 coding gene and the S. enterica genome are downloaded from NCBI Entrez.
# Code source: Patrick Kunzmann
# License: BSD 3 clause
import matplotlib.pyplot as plt
import numpy as np
from matplotlib.collections import LineCollection
import biotite
import biotite.application.viennarna as viennarna
import biotite.database.entrez as entrez
import biotite.sequence.align as align
import biotite.sequence.graphics as seqgraphics
import biotite.sequence.io.genbank as gb
# Download Escherichia coli BL21 and Salmonella enterica genome
gb_file = gb.MultiFile.read(
entrez.fetch_single_file(["CP001509", "CP019649"], None, "nuccore", "gb")
)
ec_file, se_file = tuple(gb_file)
annot_seq = gb.get_annotated_sequence(ec_file, include_only=["ncRNA"])
# Find M1 gene in E. coli genome via its annotation
for feature in annot_seq.annotation:
if "product" in feature.qual and "RNase P" in feature.qual["product"]:
m1_sequence = annot_seq[feature]
# Get S. enterica genome sequence
se_genome = gb.get_sequence(se_file)
# We want to search in the genome sequence and its reverse complement
genomic_seqs = [se_genome, se_genome.reverse().complement()]
In an initial fast matching step, we look for matching k-mers
between M1 and the S. enterica genome.
A matching k-mer is a length k subsequence, that appears in both
sequences.
In Biotite this task is carried out by a KmerTable.
Later we will perform costly gapped alignments at the match positions. To reduce the number of triggered gapped alignments at unspecific matches, we add some additional filters: One extra condition is that two non-overlapping matches must appear on the same diagonal \(D = j - i\), where i and j are the sequence positions in the match. Furthermore, only one match is considered in a defined range of diagonals, as small deviations from the diagonal appear due to indels. Without this filter, multiple gapped alignments would be triggered in basically the same region, leading to redundant alignments.
K = 12
MAX_MATCH_DISTANCE = 20
DISCARD_RANGE = 50
table = align.KmerTable.from_sequences(K, genomic_seqs)
matches = table.match(m1_sequence)
fig = plt.figure(figsize=(8.0, 8.0))
trigger_matches = []
# 0 represents the original genome sequence, 1 the reverse complement
for strand in (0, 1):
matches_for_strand = matches[matches[:, 1] == strand]
# Plot match positions
ax = fig.add_subplot(1, 2, strand + 1)
ax.scatter(
matches_for_strand[:, 0],
matches_for_strand[:, 2] / 1e6,
s=4,
marker="o",
color=biotite.colors["dimorange"],
)
ax.set_xlim(0, len(m1_sequence))
ax.set_ylim(0, len(se_genome) / 1e6)
ax.set_xlabel("E. coli M1 position (b)")
if strand == 0:
ax.set_ylabel("S. enterica genome position (Mb)")
else: # strand == 1
ax.set_ylabel("S. enterica genome position (Mb) (reverse complement)")
# Check if there are two adjacent matches on the same diagonal
diagonals = matches_for_strand[:, 2] - matches_for_strand[:, 0]
unique_diag = np.unique(diagonals)
trigger_diagonals = np.array([], dtype=int)
for diag in unique_diag:
if np.any(np.abs(trigger_diagonals - diag) < DISCARD_RANGE):
# Discard the matches for this diagonal if this diagonal
# is too close to a diagonal that already triggers
# a gapped alignment
continue
matches_for_diagonal = matches_for_strand[diagonals == diag]
m1_positions = matches_for_diagonal[:, 0]
for pos in m1_positions:
distances = m1_positions - pos
# The other match on the same diagonal should not overlap
# with this match and should be within a cutoff range
if np.any((distances > K) & (distances < DISCARD_RANGE)):
trigger_matches.append((strand, pos, pos + diag))
trigger_diagonals = np.append(trigger_diagonals, diag)
# Only add one match per diagonal at maximum
break
print("Matches that trigger gapped alignment:")
print(trigger_matches)
fig.tight_layout()
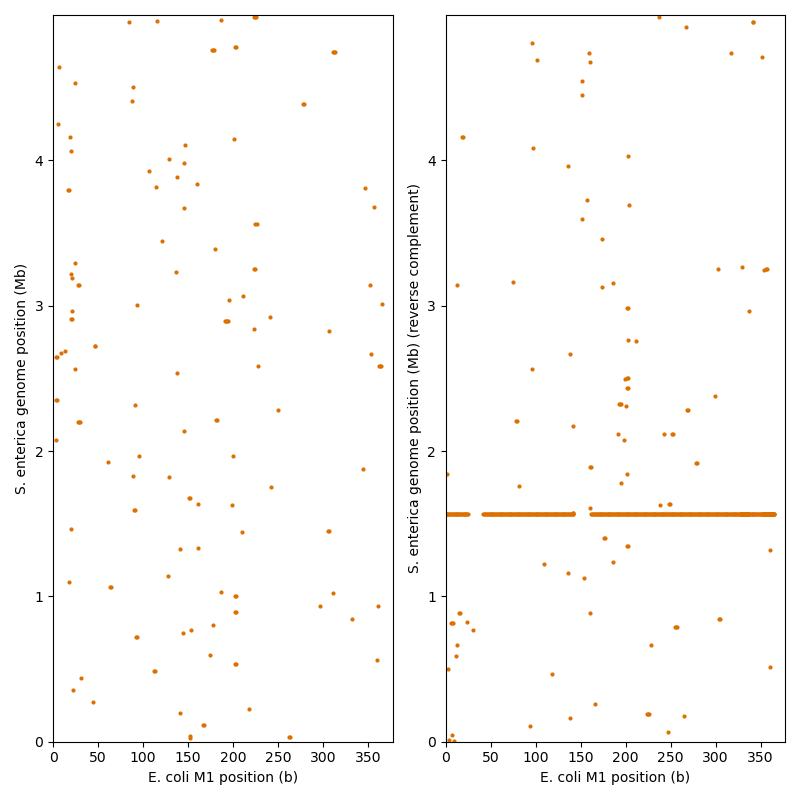
Matches that trigger gapped alignment:
[(1, np.int64(42), np.int64(1563462))]
From hundreds of initial matches, only a single one remains in this case. The diagonal of this match can be seen in the figure: It is the almost continuous line on the right side.
For the gapped alignment we use align_banded(), which reduces
the alignment search space to a narrow diagonal band.
BAND_WIDTH = 1000
alignments = []
matrix = align.SubstitutionMatrix.std_nucleotide_matrix()
for strand, m1_pos, genome_pos in trigger_matches:
genome = genomic_seqs[strand]
diagonal = genome_pos - m1_pos
alignment = align.align_banded(
m1_sequence,
genome,
matrix,
band=(diagonal - BAND_WIDTH, diagonal + BAND_WIDTH),
max_number=1,
)[0]
alignments.append((strand, alignment))
strand, best_alignment = max(
alignments, key=lambda strand_alignment: alignment[1].score
)
For visualization purposes we have to apply a renumbering function for the genomic sequence, since the indices in the alignment trace refer to the reverse complement sequence, but we want the numbers to refer to the original genomic sequence.
# Reverse sequence numbering for second sequence (genome) in alignment
number_funcs = [None, lambda x: len(best_alignment.sequences[1]) - x]
# Visualize alignment, use custom color
fig = plt.figure(figsize=(8.0, 4.0))
ax = fig.add_subplot(111)
seqgraphics.plot_alignment_similarity_based(
ax,
best_alignment,
matrix=matrix,
labels=["E. coli M1 coding gene", "S. enterica genome"],
show_numbers=True,
number_functions=number_funcs,
show_line_position=True,
color=biotite.colors["brightorange"],
)
fig.tight_layout()

The results show, that E. coli and S. enterica M1 are almost identical.
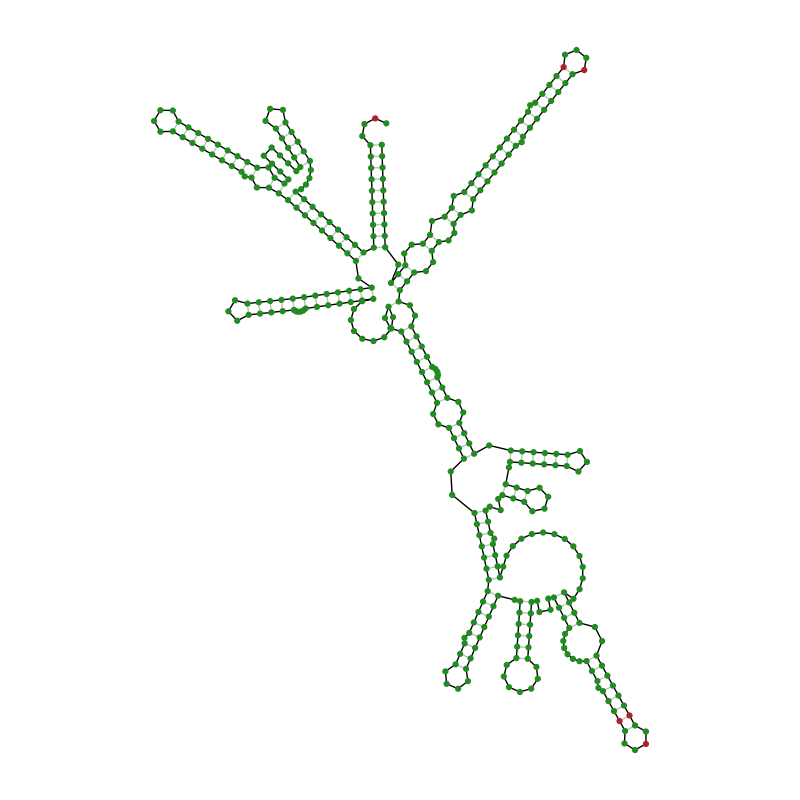
Finally, we predict and plot the secondary structure of the M1 RNA with help from ViennaRNA and highlight mismatch position between E. coli and S. enterica M1.
app = viennarna.RNAfoldApp(m1_sequence)
app.start()
app.join()
base_pairs = app.get_base_pairs()
app = viennarna.RNAplotApp(base_pairs=base_pairs, length=len(m1_sequence))
app.start()
app.join()
plot_coord = app.get_coordinates()
codes = align.get_codes(best_alignment)
m1_no_gap_codes = codes[codes[:, 0] != -1]
identities = m1_no_gap_codes[0] == m1_no_gap_codes[1]
fig = plt.figure(figsize=(8.0, 8.0))
ax = fig.add_subplot(111)
# Plot base connections
ax.plot(*plot_coord.T, color="black", linewidth=1, zorder=1)
# Plot base pairings
ax.add_collection(
LineCollection(
[(plot_coord[i], plot_coord[j]) for i, j in base_pairs],
color="silver",
linewidth=1,
zorder=1,
)
)
# Plot base markers
ax.scatter(
*plot_coord.T,
s=12,
# Render markers over lines
zorder=2,
# Display base marker color based on the identity in the alignment
color=["forestgreen" if identity else "firebrick" for identity in identities],
)
ax.set_aspect("equal")
ax.axis("off")
fig.tight_layout()
plt.show()