Note
Go to the end to download the full example code
Quantifying gene expression from RNA-seq data#
This example demonstrates how Biotite’s alignment functionalities can be used to map reads from RNA sequencing to a genome (or more precisely to cDNA data). This enables counting the number of transcripts for each gene in the expression data. These raw counts can be used downstream for transcriptomics analyses, which is out of scope of Biotite.
Note
The approach shown here shows only the backbone of a read mapper. For writing an actual program, this example should be extended, e.g. with more precise acceptance criteria for alignments.
# Code source: Patrick Kunzmann
# License: BSD 3 clause
import functools
import gzip
import multiprocessing
from io import StringIO
import matplotlib.pyplot as plt
import numpy as np
import pandas as pd
import requests
import biotite
import biotite.application.sra as sra
import biotite.sequence as seq
import biotite.sequence.align as align
import biotite.sequence.io.fasta as fasta
import biotite.sequence.io.fastq as fastq
# The number of processes for read mapping
N_PROCESS = 2
# This example script is only a demonstration
# -> decrease number of processed reads to decrease its run time
EXCERPT = 100000
# A flat Phred quality score threshold under which a read is ignored
QUALITY_THRESHOLD = 30
# k-mer length for matching reads to genes
K = 15
# Window length of minimizers
WINDOW = 10
# Band width for banded gapped alignment
BAND_WIDTH = 10
# Number of highest expressed genes to display
N_TOP_LIST = 20
# URL of the cDNA for the genome of interest
CDNA_URL = (
"https://ftp.ensemblgenomes.ebi.ac.uk/pub/plants/release-57/fasta/"
"arabidopsis_thaliana/cdna/Arabidopsis_thaliana.TAIR10.cdna.all.fa.gz"
)
# SRA UID for the RNA-seq data
READS_UID = "SRR6919890"
Fetching the data#
For the purpose of this example expression data from the plant Arabidopsis thaliana is analyzed. The sequence reads are downloaded from the NCBI Sequence Read Archive (SRA).
app = sra.FastqDumpApp(READS_UID)
app.start()
app.join()
# Single-ended -> Only one FASTQ
fastq_path = app.get_file_paths()[0]
To quantify the expression from the RNA-seq data, we need a reference, to which the reads can be aligned to. Since RNA-seq reads are at hand a fitting reference are the transcript sequences (cDNA) of the genome from the species, the RNA-seq data was recorded from.
We could have generated the cDNA sequences ourselves by reading the
gene coordinates from a GFF file
(via biotite.sequence.io.gff.GFFFile) and extracting the
corresponding sequences from the genome sequence FASTA file
(via biotite.sequence.io.fasta.FastaFile).
However, to keep this example more focussed, the precomputed cDNA
sequences are simply fetched from EnsemblPlants
[1].
The following code reads each entry in the cDNA FASTA file and extracts the gene symbols, i.e. the ‘names’ of the genes, and the corresponding cDNA sequences.
def get_gene_symbol(header):
fields = header.split()
for field in fields:
if field.startswith("gene_symbol:"):
# Get only the actual gene symbol
# -> remove 'gene_symbol:' prefix
return field.replace("gene_symbol:", "")
# No gene symbol for this cDNA (e.g. non-coding)
return None
response = requests.get(CDNA_URL)
fasta_content = gzip.decompress(response.content).decode("UTF-8")
gene_symbols = []
sequences = []
for header, seq_string in fasta.FastaFile.read_iter(StringIO(fasta_content)):
symbol = get_gene_symbol(header)
if symbol:
# Check if the length is large enough to be used in the
# k-mer table (see below)
if len(seq_string) < K + WINDOW - 1:
print(f"Ignored {symbol}: Cannot be indexed")
continue
gene_symbols.append(symbol)
try:
# Use the unambiguous alphabet (ACGT) to increase the length
# of k-mers later on:
# The k-mer code in restricted to int64, so a larger number
# of base alphabet codes decreases the *k* that fits into
# the integer type
sequences.append(seq.NucleotideSequence(seq_string, ambiguous=False))
except seq.AlphabetError:
# For the simplicity of this example just ignore sequences
# with unambiguous symbols
# This applies only to a few cDNA sequences
print(f"Ignored {symbol}: Contains ambiguous symbol")
print("\nExcerpt of genes:\n")
for symbol in gene_symbols[:20]:
print(symbol)
Ignored AT4G33735: Cannot be indexed
Ignored AT2G08986: Contains ambiguous symbol
Ignored ORC4: Contains ambiguous symbol
Ignored ORC4: Contains ambiguous symbol
Ignored AT5G24575: Cannot be indexed
Ignored REF4: Contains ambiguous symbol
Ignored AT1G33612: Contains ambiguous symbol
Excerpt of genes:
AER
AT4G32100
AT2G43120
AT2G43120
AT1G30814
AT1G30814
AT1G30814
AT1G30814
PUB29
PUM6
PUM6
PUM6
PUM6
PUM6
AT1G78930
AT3G07950
AT4G03450
AT4G03450
AT1G71695
AT1G58983
Aligning reads to the reference#
The final aim is to obtain for each read an alignment to the cDNA sequence, i.e. the gene, it originates from. However, performing the usual sequence alignment of each read to every cDNA sequence would be computationally infeasible. Instead a fast matching step is performed first to select only those cDNA sequences that probably would give a high-scoring alignment for a read.
Here the general approach from the read mapper Minimap
[2] is adopted:
The cDNA sequences are first decomposed into k-mers.
Then the minimizers are chosen from the k-mers
[3].
In short, the minimizer is the smallest k-mer in a running window
of k-mers.
The effect is that the number of k-mers to be matched against later
is drastically reduced, while the sensitivity of finding a match with
the correct cDNA is still good, if they are highly similar.
This assumption only holds, if sequencing is conducted with high
fidelity.
For fast matching the minimizers of all cDNA sequences are indexed
into a BucketKmerTable, the memory-efficient twin of the
KmerTable:
While a :class:`KmerTable would require $4^k approx$ 1 billion
buckets (one for each k-mer), the class:BucketKmerTable limits the
number of buckets, but requires a bit more computation time for its
construction and matching.
base_alph = seq.NucleotideSequence.alphabet_unamb
kmer_alph = align.KmerAlphabet(base_alph, K)
min_selector = align.MinimizerSelector(kmer_alph, WINDOW, align.RandomPermutation())
kmer_table = align.BucketKmerTable.from_kmer_selection(
kmer_alph, *zip(*[min_selector.select(sequence) for sequence in sequences])
)
Now the reads can be matched to the indexed cDNA sequences. For this purpose they need to be processed in the same way: They are decomposed into k-mers and the minimizers are selected. After matches to certain cDNA sequences have been identified, the read is aligned to each of these sequences. As noted before, this script assumes sequencing was performed with high fidelity. Thus, the expected probability of indels is relatively small. This circumstance can be leveraged to decrease the computation time even further: Instead of allowing an arbitrary number of gaps between the read and the cDNA, the number of gaps is restricted [4]. Hence, not the entire alignment space is explored, but only a thin band. The required width of the band can be computed based on the indel probability [5], but for the sake of brevity a flat constant is used here. After all alignments have been collected, simply the highest-scoring one is chosen as the correct one.
def map_read(read_string, kmer_table, gene_sequences, substitution_matrix):
try:
read = seq.NucleotideSequence(read_string, ambiguous=False)
except seq.AlphabetError:
# There are a few reads that may contain unambiguous symbols
# For the same reason as explained above, these are ignored
return
# Fast matching of minimizers
matches = kmer_table.match_kmer_selection(*min_selector.select(read))
if len(matches) == 0:
# No matching gene found for read
return
# The probability that a read matches a gene at two different
# positions that would give distinct alignments is tiny
# -> For each gene take only the first matched position
matched_gene_indices, indices = np.unique(matches[:, 1], return_index=True)
matched_diagonals = matches[indices, 2] - matches[indices, 0]
# For each matched gene perform a more thorough gapped alignment
alignments = [
(
gene_i,
align.align_banded(
read,
gene_sequences[gene_i],
substitution_matrix,
band=(diagonal - BAND_WIDTH, diagonal + BAND_WIDTH),
gap_penalty=-10,
max_number=1,
)[0],
)
for gene_i, diagonal in zip(matched_gene_indices, matched_diagonals)
]
# We assume that the best alignment is the correct one
gene_index, alignment = max(alignments, key=lambda ali: ali[1].score)
return gene_index, alignment
Let’s perform a read mapping for a single read.
substitution_matrix = align.SubstitutionMatrix.std_nucleotide_matrix()
for i, (_, (seq_string, q)) in enumerate(
fastq.FastqFile.read_iter(fastq_path, offset="Sanger")
):
# For demonstration only a single clean read is mapped
if i == 3:
read_string = seq_string
break
gene_index, alignment = map_read(
read_string, kmer_table, sequences, substitution_matrix
)
print(f"Match: {gene_symbols[gene_index]}")
print(alignment)
Match: MYB49
GGTTGATCTAGACCAGAAAATCCAAATGGTCAAAGAACTTGATGGACATGG
GGTTGATCTAGACCAGAAAATCCAAATGGTCAAAGAACTTGATGGACATGT
For the thousands of reads that need to be mapped here, it is
reasonable to divide the work into multiple processes.
As the BucketKmerTable needs to be copied to each of the
spawned processes, a long startup time can be expected.
However, for the large number of reads which can be then processed in
parallel, it is still worth it.
def read_iter(fastq_path):
for i, (_, (read_string, quality)) in enumerate(
fastq.FastqFile.read_iter(fastq_path, offset="Sanger")
):
# For the purpose of this example only a faction of the reads
# are processed to save computation time
if i >= EXCERPT:
break
# Very simple filtering of low-quality reads
if np.mean(quality) < QUALITY_THRESHOLD:
continue
yield read_string
with multiprocessing.Pool(processes=N_PROCESS) as p:
# Use multiprocessing to map reads to genes
# and remove non-mappable reads (None values) afterwards
mapping_results = list(
filter(
lambda mapping: mapping is not None,
p.map(
functools.partial(
map_read,
kmer_table=kmer_table,
gene_sequences=sequences,
substitution_matrix=substitution_matrix,
),
read_iter(fastq_path),
),
)
)
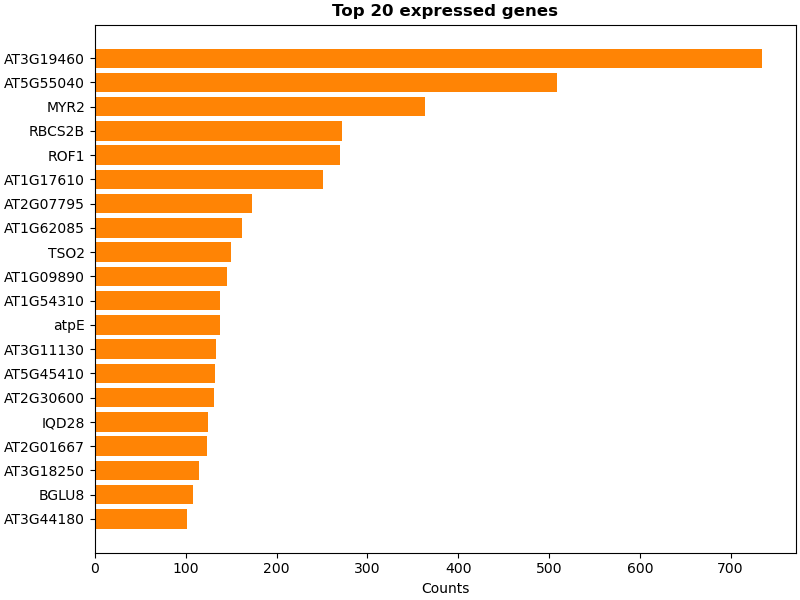
Now the genes are counted: For each read, the count of the gene corresponding to the aligned cDNA is incremented. These counts can be used as input for further analysis transcriptomics pipelines. For the scope of this example simply the most abundant genes are displayed. The alignment itself is also discarded here, but note that it could also be used in downstream analysis.
Read alignments are typically stored in file formats like SAM/BAM
[6].
The package pysam
provides an interface to these formats.
To convert an alignment into a CIGAR string, the function
write_alignment_to_cigar() can be used.
counts = np.zeros(len(sequences), dtype=int)
for gene_index, alignment in mapping_results:
counts[gene_index] += 1
# Show most expressed genes first
order = np.argsort(counts)[::-1]
ranked_gene_symbols = [gene_symbols[i] for i in order]
ranked_counts = counts[order]
# Put into dataframe for prettier printing
counts = pd.DataFrame(
{"gene_symbol": ranked_gene_symbols, "count": ranked_counts},
index=np.arange(1, len(ranked_counts) + 1),
)
# Show Top N
top_counts = counts[:N_TOP_LIST]
top_counts
Finally the top expressed genes are plotted.
figure, ax = plt.subplots(figsize=(8.0, 6.0), constrained_layout=True)
ax.barh(top_counts["gene_symbol"], top_counts["count"], color=biotite.colors["orange"])
ax.invert_yaxis()
ax.set_title(f"Top {N_TOP_LIST} expressed genes", weight="semibold")
ax.set_xlabel("Counts")
plt.show()