Note
Go to the end to download the full example code
Identification of a binding site by sequence conservation#
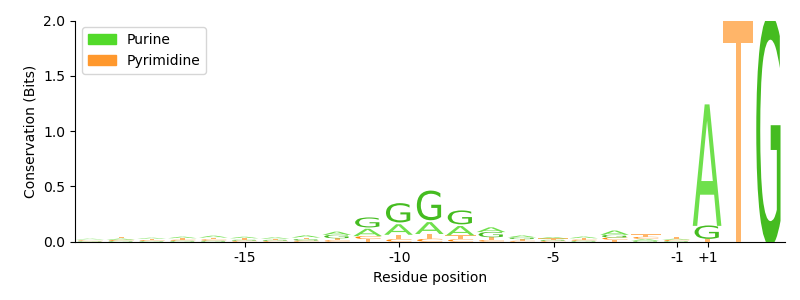
In this example we identify the ribosomal binding site on mRNA, also called Shine-Dalgarno sequence, in Escherichia coli.
In the beginning of the translation the 16S rRNA of the ribosome recognizes this purine-rich region on the mRNA, which typically lies a few bases upstream of the start codon. After binding the sequence, the ribosome starts scanning the mRNA in downstream direction to locate the start codon.
# Code source: Patrick Kunzmann
# License: BSD 3 clause
import tempfile
import matplotlib.pyplot as plt
import matplotlib.ticker as ticker
import numpy as np
from matplotlib.patches import Patch
import biotite
import biotite.database.entrez as entrez
import biotite.sequence as seq
import biotite.sequence.graphics as graphics
import biotite.sequence.io.genbank as gb
UTR_LENGTH = 20
### Get the E. coli K-12 genome as annotated sequence
gb_file = gb.GenBankFile.read(
entrez.fetch("U00096", tempfile.gettempdir(), "gb", "nuccore", "gb")
)
# We are only interested in CDS features
bl21_genome = gb.get_annotated_sequence(gb_file, include_only=["CDS"])
### Extract sequences for 5' untranslated regions (UTRs)
# In this case we define the untranslated region, as the sequence
# up to UTR_LENGTH bases upstream from the start codon
utrs = []
for cds in bl21_genome.annotation:
# Expect a single location for the feature,
# since no splicing can occur
# Ignore special cases like ribosomal slippage sites, etc.
# for simplicity
if len(cds.locs) != 1:
continue
# Get the only location for this feature
loc = list(cds.locs)[0]
# Get the region behind or before the CDS, based on the strand the
# CDS is on
if loc.strand == seq.Location.Strand.FORWARD:
utr_start = loc.first - UTR_LENGTH
utr_stop = loc.first
# Include the start codon (3 bases) in the UTRs for later
# visualization
utrs.append(bl21_genome[utr_start : utr_stop + 3].sequence)
else:
utr_start = loc.last + 1
utr_stop = loc.last + 1 + UTR_LENGTH
utrs.append(
bl21_genome[utr_start - 3 : utr_stop].sequence.reverse().complement()
)
### Create profile
# Increase the counter for each base and position
# while iterating over the sequences
frequencies = np.zeros((UTR_LENGTH + 3, len(bl21_genome.sequence.alphabet)), dtype=int)
for utr in utrs:
frequencies[np.arange(len(utr)), utr.code] += 1
profile = seq.SequenceProfile(
symbols=frequencies,
gaps=np.zeros(len(frequencies)),
alphabet=bl21_genome.sequence.alphabet,
)
### Visualize the profile
# Spend extra effort for correct sequence postion labels
def normalize_seq_pos(x):
"""
Normalize sequence position, so that the position of the upstream bases is negative.
"""
# Sequence positions are always integers
x = int(x)
x -= UTR_LENGTH
# There is no '0' position
if x <= 0:
x -= 1
return x
@ticker.FuncFormatter
def sequence_loc_formatter(x, pos):
x = normalize_seq_pos(x)
return f"{x:+}"
COLOR_SCHEME = [
biotite.colors["lightgreen"], # A
biotite.colors["orange"], # C
biotite.colors["dimgreen"], # G
biotite.colors["brightorange"], # T
]
fig, ax = plt.subplots(figsize=(8.0, 3.0))
graphics.plot_sequence_logo(ax, profile, COLOR_SCHEME)
normalized_pos = np.array([normalize_seq_pos(x) for x in range(len(profile.symbols))])
tick_locs = np.where(np.isin(normalized_pos, [-15, -10, -5, -1, 1]))[0]
ax.set_xticks(tick_locs)
ax.xaxis.set_major_formatter(ticker.FuncFormatter(sequence_loc_formatter))
ax.set_xlabel("Residue position")
ax.set_ylabel("Conservation (Bits)")
ax.spines["right"].set_visible(False)
ax.spines["top"].set_visible(False)
ax.legend(
loc="upper left",
handles=[
Patch(color=biotite.colors["green"], label="Purine"),
Patch(color=biotite.colors["lightorange"], label="Pyrimidine"),
],
)
fig.tight_layout()
plt.show()