Note
Go to the end to download the full example code
Detection of disulfide bonds#
This example presents a function that detects disulfide bridges
in protein structures.
Then the detected disulfide bonds are visualized and added to the
bonds attribute of the AtomArray.
The employed criteria for disulfide bonds are quite simple in this case: the \(S_\gamma\) atoms of two cystein residues must be in a vicinity of \(2.05 \pm 0.05\) Å and the dihedral angle of \(C_\beta - S_\gamma - S^\prime_\gamma - C^\prime_\beta\) must be \(90 \pm 10 ^{\circ}\).
# Code source: Patrick Kunzmann
# License: BSD 3 clause
import io
from tempfile import gettempdir
import matplotlib.patches as patches
import matplotlib.pyplot as plt
import numpy as np
import biotite.database.rcsb as rcsb
import biotite.sequence as seq
import biotite.structure as struc
import biotite.structure.io.pdbx as pdbx
def detect_disulfide_bonds(
structure, distance=2.05, distance_tol=0.05, dihedral=90, dihedral_tol=10
):
# Array where detected disulfide bonds are stored
disulfide_bonds = []
# A mask that selects only S-gamma atoms of cysteins
sulfide_mask = (structure.res_name == "CYS") & (structure.atom_name == "SG")
# sulfides in adjacency to other sulfides are detected in an
# efficient manner via a cell list
cell_list = struc.CellList(
structure, cell_size=distance + distance_tol, selection=sulfide_mask
)
# Iterate over every index corresponding to an S-gamma atom
for sulfide_i in np.where(sulfide_mask)[0]:
# Find indices corresponding to other S-gamma atoms,
# that are adjacent to the position of structure[sulfide_i]
# We use the faster 'get_atoms_in_cells()' instead of
# `get_atoms()`, as precise distance measurement is done
# afterwards anyway
potential_bond_partner_indices = cell_list.get_atoms_in_cells(
coord=structure.coord[sulfide_i]
)
# Iterate over every index corresponding to an S-gamma atom
# as bond partner
for sulfide_j in potential_bond_partner_indices:
if sulfide_i == sulfide_j:
# A sulfide cannot create a bond with itself:
continue
# Create 'Atom' instances
# of the potentially bonds S-gamma atoms
sg1 = structure[sulfide_i]
sg2 = structure[sulfide_j]
# For dihedral angle measurement the corresponding
# C-beta atoms are required, too
cb1 = structure[
(structure.chain_id == sg1.chain_id)
& (structure.res_id == sg1.res_id)
& (structure.atom_name == "CB")
]
cb2 = structure[
(structure.chain_id == sg2.chain_id)
& (structure.res_id == sg2.res_id)
& (structure.atom_name == "CB")
]
# Measure distance and dihedral angle and check criteria
bond_dist = struc.distance(sg1, sg2)
bond_dihed = np.abs(np.rad2deg(struc.dihedral(cb1, sg1, sg2, cb2)))
if (
bond_dist > distance - distance_tol
and bond_dist < distance + distance_tol
and bond_dihed > dihedral - dihedral_tol
and bond_dihed < dihedral + dihedral_tol
):
# Atom meet criteria -> we found a disulfide bond
# -> the indices of the bond S-gamma atoms
# are put into a tuple with the lower index first
bond_tuple = sorted((sulfide_i, sulfide_j))
# Add bond to list of bonds, but each bond only once
if bond_tuple not in disulfide_bonds:
disulfide_bonds.append(bond_tuple)
return np.array(disulfide_bonds, dtype=int)
As test case a structure of a cysteine knot protein is used, specifically the squash trypsin inhibitor EETI-II (PDB: 2IT7). This motif is famous for its three characteristic disulfide bridges forming a ‘knot’. However, the loaded PDBx file already has information about the the disulfide bridges. To have a proper test case, all disulfide bonds are removed from the structure and we pretend that the structure never had information about the disulfide bonds. For later verification that the implemented function works correctly, the disulfide bonds, that are removed, are printed out.
pdbx_file = pdbx.BinaryCIFFile.read(rcsb.fetch("2IT7", "bcif", gettempdir()))
knottin = pdbx.get_structure(pdbx_file, include_bonds=True, model=1)
sulfide_indices = np.where((knottin.res_name == "CYS") & (knottin.atom_name == "SG"))[0]
for i, j, _ in knottin.bonds.as_array():
if i in sulfide_indices and j in sulfide_indices:
print(knottin[i])
print(knottin[j])
print()
knottin.bonds.remove_bond(i, j)
A 2 CYS SG S -5.182 1.760 -2.385
A 19 CYS SG S -4.800 -0.230 -2.187
A 9 CYS SG S 4.169 -0.459 -0.194
A 21 CYS SG S 5.347 -0.755 -1.829
A 15 CYS SG S 0.741 -1.839 -1.408
A 27 CYS SG S 0.436 -1.062 0.449
Now the sanitized structure is put into the disulfide detection function. The detected bonds are printed out and we expect to see the same bonds, that were removed in the code snippet above.
disulfide_bonds = detect_disulfide_bonds(knottin)
for sg1_index, sg2_index in disulfide_bonds:
print(knottin[sg1_index])
print(knottin[sg2_index])
print()
A 2 CYS SG S -5.182 1.760 -2.385
A 19 CYS SG S -4.800 -0.230 -2.187
A 9 CYS SG S 4.169 -0.459 -0.194
A 21 CYS SG S 5.347 -0.755 -1.829
A 15 CYS SG S 0.741 -1.839 -1.408
A 27 CYS SG S 0.436 -1.062 0.449
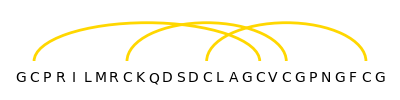
The found disulfide bonds are visualized with the help of Matplotlib: The amino acid sequence is written on the X-axis and the disulfide bonds are depicted by yellow semi-ellipses.
# Create a sequence object for each residue in the structure
# As we want each residue to appear only once in the sequence,
# we choose an atom that appears in each residue once: the CA
sequence = seq.ProteinSequence(knottin.res_name[knottin.atom_name == "CA"])
figure = plt.figure(figsize=(4.0, 1.0))
ax = figure.gca()
MARGIN = 0.2
ax.set_xlim(1 - MARGIN, len(sequence) + MARGIN)
ax.set_ylim(0, 1 + MARGIN)
ax.set_xticks(np.arange(1, len(sequence) + 1))
ax.set_xticklabels(str(sequence))
ax.yaxis.set_tick_params(left=False, right=False, labelleft=False, labelright=False)
ax.xaxis.set_tick_params(
bottom=True, top=False, labelbottom=True, labeltop=False, width=0
)
ax.set_frame_on(False)
for sg1_index, sg2_index in disulfide_bonds:
sg1_res_id = knottin.res_id[sg1_index]
sg2_res_id = knottin.res_id[sg2_index]
ellipse_center = (sg1_res_id + sg2_res_id) / 2
ellipse_width = sg2_res_id - sg1_res_id
# Height is 2 instead of 1,
# because only the upper half of the ellipse is visible
ax.add_patch(
patches.Ellipse(
xy=(ellipse_center, 0),
width=ellipse_width,
height=2,
facecolor="None",
edgecolor="gold",
linewidth=2,
)
)
figure.tight_layout()
Finally, the detected bonds are added to the structure. Basically, the removal step above is reversed.
for sg1_index, sg2_index in disulfide_bonds:
knottin.bonds.add_bond(sg1_index, sg2_index, struc.BondType.SINGLE)
# The structure with added disulfide bonds
# could now be written back into a structure file
#
out_file = pdbx.BinaryCIFFile()
pdbx.set_structure(out_file, knottin)
out_file.write(io.BytesIO())
plt.show()